

Primary Biliary Cholangitis (PBC)
Primary biliary cholangitis (PBC) is a cholestatic, slowly progressive, autoimmune liver disease affecting the small biliary ducts within the liver. PBC can be diagnosed anytime during adulthood but the majority of patients are diagnosed between the ages 30 and 60. Females are predominately affected and account for 90% of all cases. In fact, the female-to-male ratio is 9-22 : 1.
The etiology of PBC is unknown, but is presumed to be a combination of genetic predisposition and environmental exposures that trigger the immune system to fight the small bile ducts within the liver. This leads to problems with bile removal from the liver leading to buildup causing chronic inflammation and damage and eventually leading to cirrhosis. Other autoimmune conditions occur more frequently in patients with PBC, including thyroid disorders, Raynaud syndrome, rheumatoid arthritis and Sjogren’s syndrome. Fifteen percent of those with PBC may develop an overlap syndrome with autoimmune hepatitis in which the immune system also targets hepatocytes (liver cells).
The hallmark of the disease is a positive anti-mitochondrial antibodies (AMA) test, which is found in 95% of the cases. The liver test most affected in PBC is the alkaline phosphatase. The AST/ALT levels are typically only mildly elevated and the serum bilirubin is usually normal in the early stages of the disease. Other labs that can be elevated include IgM and cholesterol levels though this does not directly relate to increased risk of atherosclerosis or heart disease.
PBC is diagnosed provided two of the following three criteria are met: (1) AMA positive (2) alkaline phosphatase (AP) > 1.5 times the upper limit of normal x 6 mo; and (3) compatible liver biopsy (Figure 1). Patients who lack AMA but whose clinical presentation, labs and biopsy are consistent with PBC, are deemed to have AMA-negative PBC. At the earliest stage of the disease, individuals can have a positive AMA and minimal abnormalities. Although liver biopsy is not mandatory for diagnosis, it can help to stage the disease and differentiate PBC from other cholestatic liver disorders, if these are on the differential. PBC has 4 histological stages (Table 1), which occur sequentially with disease progression.
The most common presenting symptom of PBC is itching which is worse at night however fatigue is generally present in most patients when asked. Signs of PBC may not be present at all however could include skin darkening, enlargement of the liver or spleen and xanthelasma (cholesterol deposits). Yellowing of the eyes or skin occurs late in the course of the disease. Individuals often develop vitamin deficiencies (Vitamin A, D, E, and K) and premature thinning of the bones.
Medical therapy for PBC are limited. Steroids do not alter the course of the disease and can worsen or accelerate the onset of bone loss. Ursodeoxycholic acid (UDCA), has been shown to improve transplant-free survival and delay time to transplant. The recommended dose of UDCA is 13-15 mg/kg/day and is well tolerated. Improvements in liver tests are usually seen within the first 6-9 months. However, up to 40% of PBC patients treated with UDCA do not respond. These patients are at risk of disease progression.
Another therapy that has recently been FDA approved is Obeticholic acid (OCA), a novel farnesoid-X-receptor (FXR) agonist. It works by inhibiting bile acid production and increasing the removal of bile acids from the liver cells. A starting dose of 5 mg daily increased to 10 mg based on response is recommended. The most common side effect from OCA is itching and mild triglyceride elevation. The dose for decompensated cirrhosis is 5 mg weekly.
Important to patients with PBC, is the management of the symptoms. Itching is a challenging complaint. Cholestyramine is commonly used to treat this symptom with variable success. Topical treatments and lotions to keep skin from drying out may be beneficial. Other therapies with varying results include hydroxyzine, Benadryl, sertraline, phenobarbital, rifampin, and naloxone. Long-term management of patients with PBC include: liver tests every 3-6 months, yearly evaluation for thyroid illness (TSH), bone mineral densitometry every 2-4 years, and assessment for fat-soluble vitamin deficiencies yearly if the bilirubin is >2.0.
Ultimately patients with end-stage PBC will need a liver transplantation as a life-saving treatment. Survival rates from liver transplantation are close to 90% at 1-year and 85% at 5-years. Recurrence after liver transplantation can happen but is uncommon.
Figure 1. Histological features of PBC
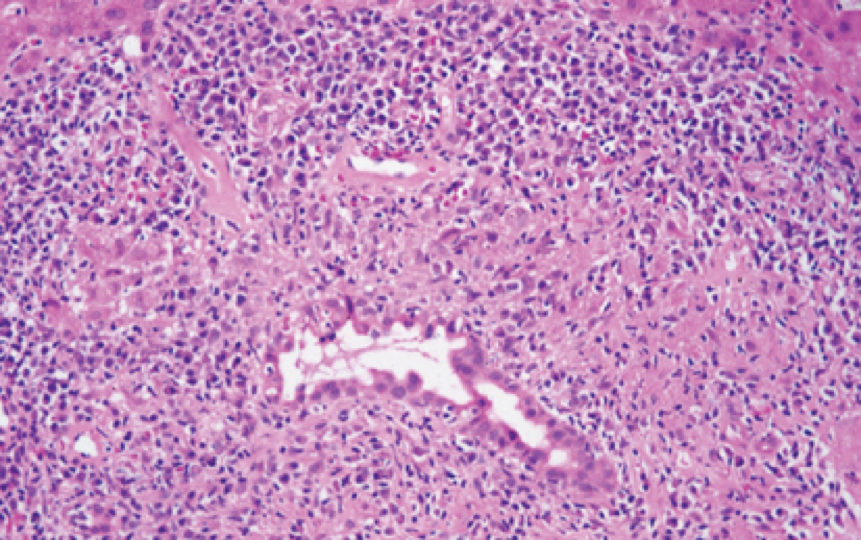
Journal of Online Hepatology. Posted on September 21, 2011. Hematoxylin and eosin stainof Portal inflammation with florid duct lesion.
Table 1. Histological Stages of PBC
Stage | Description |
Stage I | Portal inflammation with or without florid bile duct lesions (focal lesions that exhibit intense inflammatory infiltration and necrosis around bile ducts) |
Stage II | Increase in size of periportal lesions with interface hepatitis |
Stage III | Distortion of hepatic architecture with numerous fibrous septa with disease progression. |
Stage IV | Cirrhosis |
References if needed:
AASLD guidelines
Zakharia, Kais. Liver International Sept 2017 Complications, symptoms, quality of life and pregnancy in cholestatic liver disease